

Self-assembly: artist’s impression of amyloid plaques (orange) within a brain. (Courtesy: iStock/selvanegra)
I decided for my next review, to look at the potential role of ketone supplementation in people with cognitive decline/dementia, largely in the form of Alzheimer’s Disease (AD). The reasons include; it’s a very common, pathophysiology is extensively studied, there is no good treatment currently available and cerebral energetics plays a role. Furthermore, 2nd to epilepsy, it’s the most studied application of exogenous ketone supplementation in humans. As the reader will see, the data is encouraging although much more work needs to be done before a definitive recommendation can be made.
I’ve tried to make the topic as readable as possible, but there is a lot of information to cover. As always, I’ll attempt to explain difficult concepts as best I can. Other interesting reading can be found here - https://www.neuroscientificallychallenged.com/blog/know-your-brain-alzheimers-disease
So, what’s the big deal with Alzheimer’s?
Alzheimer’s Disease is the commonest cause of dementia in adults and affects approximately 47 million people globally. The incidence depends on age but doubles every 10 years after the age of 60. As such, by the age of 85, the incidence ranges between 60 – 80 per 1000 population. Managing AD places a heavy burden on patients, their families and the health care sector in general. Aside from the physical and psychological burdens, the annual cost of managing this disease is massive and can be measured in the hundreds of billions USD.
So, by all measures, AD is a big deal! Here’s some basic science!
The aetiology of AD is poorly understood. Although a genetic component certainly exists, only a small percentage of cases can be explained by familial trait. The vast majority of cases, therefore, fall into the sporadic AD category.
Genetic risk factors include age, family history (which increases risk by 30%) and mutations affecting amyloid (an abnormal protein) beta (a type of amyloid) protein production and the presence of apolipoprotein E-ɛ4. The apolipoprotein gene is located on chromosome 19 and exists in 3 alleles (an allele is a variant form of a gene). Individuals with 2 copies of the ɛ4 allele are at most risk.
Early onset AD while responsible for only 1% AD cases, carries a much higher genetic burden then late onset. In late onset disease, the most firmly established genetic link is related to the presence of the APOE- ɛ4 allele which leads to a 3-fold increase risk in carriers of one allele, or 12 times for carriers of two alleles.
For the majority of sporadic AD sufferers who do not have a defined genetic predisposition, risk factors include hypertension, chronic vascular disease, dyslipidaemia, obesity, diabetes, lack of exercise and traumatic brain injury. Certain medications are also implicated including benzodiazepines, anticholinergics and proton pump inhibitors. Certain environmental risk factors have been identified including smoking, air pollution and certain pesticides.
How does AD present?

Classically, memory impairment is the most common initial symptom of AD. The pattern is distinctive ie. Initially declarative memory goes– unable to remember events in terms of time and place. Semantic memory (ability to remember facts such as vocabulary and concepts) tends to deteriorate later in the disease. The initial memory deficit is best described as anterograde long-term episodic amnesia (although long term may describe events anywhere from minutes to years).
Other clinical problems include behavioural abnormalities, loss of executive function, sleep disorders, apraxia (difficulty with skilled movement), seizures and loss of motor functions.
AD is relentless and leads to progressive deterioration with the average life expectancy 8-10 years after diagnosis.
Can I diagnose AD prior to clinical symptoms?
Imaging – as I general rule, imaging is more of a research tool and used to exclude other pathologies.
MRI scan show nonspecific atrophy- characteristically in region of the hippocampus and medial temporal lobe of the brain.
Functional brain imaging with 18-F fluorodeoxyglucose positron emission tomography (FDG-PET) or single-photon emission computed tomography (SPECT) demonstrates cerebral hypo-perfusion and hypo-metabolism.
Aside – this is a very interesting finding and I’ll return to the topic later.
Experimental imaging includes Amyloid PET tracers measuring the amyloid lesion burden and Tau PET tracers. (Both still undergoing research)
What about blood tests?
Again, largely experimental. A decrease in Aβ42 and increase in total tau concentrations in cerebrospinal fluid. (This requires a lumbar puncture so not really practical. Also, these findings are suggestive of AD not diagnostic)
Diagnosis (post-morbid – refers to diagnosis once symptoms begin)
(only way to diagnose pre-morbidly is via brain biopsy)
As no test is definitive, the diagnosis is based on criteria established by the National Institute on Aging and the Alzheimers Association or the DSM 5.
Once the diagnosis has been made, what are the treatment options?
Unfortunately, there is no cure for AD. Treatment is mainly symptomatic i.e. managing behavioural disturbances, environmental manipulations and counselling.
There is some evidence that treatment with cholinesterase inhibitors (e.g. Donepezil) or NMDA receptor antagonists (memantine) may slow deterioration.
Trials with antioxidants have recorded mixed results. Vitamin E (alpha tocopherol) and selegiline (a monoamine oxidase inhibitor) have both been studied in AD. Overall, vitamin E may confer a modest benefit in delaying functional progression although this is difficult to measure. There does not appear to be an advantage in selegiline which is more costly.
Unproven therapies include things like estrogen replacement therapy, NSAID, Ginkgo biloba, statins and general vitamin replacement
Pathology – what is seen under a microscope
There appears to be 3 stages in the pathology of AD – pre-clinical or asymptomatic, mild cognitive impairment (MCI) and dementia.
Pathological features include; (the list is purely for completeness and can be otherwise ignored)
· Extracellular amyloid plaques
· Cerebral amyloid angiopathy
· Intracellular neurofibrillary tangles (abnormally phosphorylated tau protein)
· Abnormal glial cell responses
· Neuronal and synaptic loss
· Decrease GLUT transporters

Amyloid plaques (pink) and neurofibrillary tangles (black) in Alzheimer's disease brain tissue. Source: www.alzheimers.org.uk.
Pathogenesis or how do these changes of AD develop
Amyloid cascade hypothesis – 1992 – amyloid beta in the form of amyloid plaques (deposited in the brain) disrupt cerebral function leading to dementia and eventual death. However, drugs targeting amyloid have largely proved ineffective.
Mitochondrial cascade hypothesis – 2004 – changes in mitochondrial (the energy powerhouse of the cell) function brought on by aging and metabolic stress initiates the events which lead to changes in biomarkers and cerebral dysfunction found in AD.
Changes in mitochondrial function include;
· Insulin resistance
· Decrease mitochondrial respiration
· Decrease key enzymes in glycolytic pathway
· Decrease glucose metabolism
· Impaired glucose utilization
· Increase lactate formation
· Increase reactive oxidant species and inflammatory cytokines
The cause of AD is clearly complicated and manifests as cerebral hypometabolism, mitochondrial dysfunction, inflammatory changes and oxidative stress.
Aside – this next section is extremely important and critical to the understanding how cerebral energy problems can lead to disease
An important observation in patients with AD is a relative reduction in glucose uptake and metabolism by the brain. This seems to occur early in the disease (before clinical symptoms develop) and can be demonstrated on FDG-PET as an early marker of AD, with studies showing 90% sensitivity. Why this occurs and how it relates to the development of AD is not known, however this early impairment in glucose metabolism may suggest early intervention to improve cerebral energetics could be effective in preventing disease progression.
Aside – a positron emission tomography (PET) with 2-[fluorine-18]fluoro-2-deoxy-D-glucose (FDG) or FDG-PET scanner is commonly used in evaluating malignancy but can also be employed in the investigation of neurodegenerative disease. FDG is an analog of glucose which becomes trapped in neurons allowing imaging and measurement of the cerebral metabolic rate for glucose.
Can ketone supplementation help?
Ketone bodies (KB) may provide an alternative to glucose as a cerebral energy source. (See prior postings) By bypassing glycolysis to produce acetyl coenzyme A – which is channelled into the Krebs cycle, KB can increase cerebral energy as it appears KB uptake is not impaired in AD.
Ketosis may be induced endogenously by starvation or exogenously through a ketogenic diet (KD) or ketone monoesters (KME) supplementation. Ketotherapy appears to improve AD through a number of mechanisms. The primary role of KB is to provide fuel for ATP production however, KB may influence mitochondrial function in a number of ways;
· Suppression of glutamate transport and increase GABA activity
· Decrease ROS and inflammatory mediators (NF-KB, COX-2, TNFα)
· Increase antioxidants
· Reduce brain Aβ42 levels
· Protect from amyloid β plaques
· Improve mitochondrial function
· Improve cell signalling
· Improve gut microbiome – modulate inflammation and cerebral blood flow
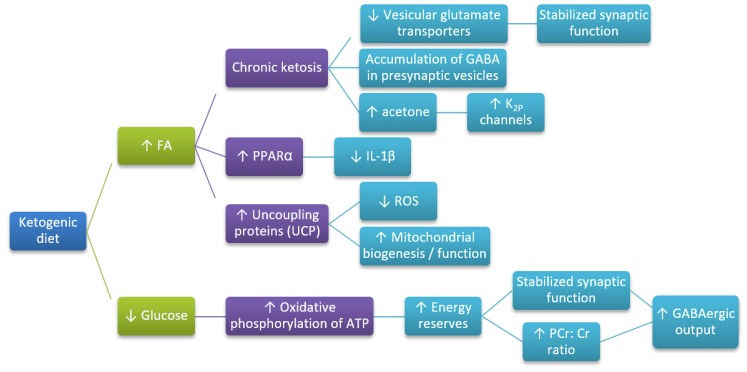
Hypothesized mechanisms through which ketogenic diet (KD) influence Alzheimer’s disease (AD) development.
(McDonald, T.J.W.; Cervenka, M.C. The expanding role of ketogenic diets in adult neurological disorders. Brain Sci. 2018, 8, 148.)
What’s the evidence from human trials?
Most studies have demonstrated an improvement in cognitive function following institution of KD. A wide range of approaches have been used including medium-chain triglycerides (MCT) diet, Mediterranean diet and use of ketone esters (KE). Correlation between clinical benefit and actual ketone concentration is difficult to demonstrate but there appears to be a dose response. Interestingly, other than minor GI disturbances, side effects are minimal. In fact, a study by Dahti et al in 2004 where obese patients were treated with 24 weeks of KD, did not find any significant side effects. Having said that, not everyone can tolerate a KD.
So, lets delve a little deeper into the evidence
In a study by Fontier (2019), 52 subjects with MCI were randomized to 30g/d MCT or placebo for 6 months. Brain ketone metabolism increased by 230% on PET scan with not change in glucose. There was significant improvement in memory, language and processing speeds. Beta-hydroxybutyrate (BHB) rose form 207 umol/l to 543 umol/l. 75% completed study. There were no serious side effects.
Neth et al (2020) compared the heart association diet (AHAD) and modified Mediterranean – ketogenic diet (MMKD) in 20 pts with MCI. MMKD improved metabolic profile and cerebral biomarkers with improvement in cerebral perfusion. Memory improved in both groups. BHB reached 700 umol/l. 90% subjects completed trial.
Aside – the diets consisted of; MMKD – 5-10% CHO, 60-65% Fats, 30% protein
AHAD – 55-65% CHO, 15-20% Fats, 23-30% protein
Costellono et al (2015) compared cerebral glucose with ketone uptake using FDG-PET scans in patients with early AD. The scan demonstrated decrease glucose metabolism whereas ketone metabolism was normal. They suggested that ketone supplementation may improve energy substrates in the AD brain.

A) Cerebral metabolic rate of acetoacetate (also a KB) (CMRa) correlates with plasma acetoacetate concentrations. plotted against fasting plasma acetoacetate (AcAc).
B) Cerebral metabolic rate of glucose (CMRg) plotted against cerebral metabolic rate of acetoacetate (CMRa). In the patients with mild AD, increase in AcAc uptake correlated with decrease in glucose uptake, suggesting a potential switch in substrate utilization.
(Castellano, Christian-Alexandre, Scott Nugent, Nancy Paquet, Sébastien Tremblay, Christian Bocti, Guy Lacombe, Hélène Imbeault, Éric Turcotte, Tamas Fulop, and Stephen C. Cunnane. “Lower Brain 18F-Fluorodeoxyglucose Uptake but Normal 11C-Acetoacetate Metabolism in Mild Alzheimer’s Disease Dementia.” Journal of Alzheimer’s Disease 43, no. 4 (2015): 1343–53.)
Aside – for me the Costellono study is critical to the understanding of the pathogenesis of dementia/delirium. The only other study I could find that looked at this aspect of delirium was by Haggstrom from 2017 who performed FDG-PET scans on 13 delirious, elderly in-patients. They found evidence of hypometabolism in all patients scanned while delirious, but this improved once the delirium settled.

FDG-PET during and after delirium. Legend: The top row (a) is the delirium scan, while the bottom row (b) is the scan taken after delirium. Darker colours indicate lower metabolism. The top row illustrates marked global hypometabolism during delirium. The bottom row illustrates an overall improvement, but not normalisation, in metabolism.
(Haggstrom, Lucy R, Julia A Nelson, Eva A Wegner, and Gideon A Caplan. “2- 18 F-Fluoro-2-Deoxyglucose Positron Emission Tomography in Delirium.” Journal of Cerebral Blood Flow & Metabolism 37, no. 11 (November 2017): 3556–67. https://doi.org/10.1177/0271678X17701764.)
A number of other studies have examined MCT supplementation (Reger et al, 2004), ketogenic formulations (Ota et al, 2019) and low versus high carbohydrate diets (Krikorian et al, 2012). Characteristically the studies ran over hours to weeks but tended to generate only mild ketosis (eg. BHB 540 umol/l – Reger et al). They all noted improvement in neurocognitive scores at the end.
Three meta-analysis including more than 10 human studies and > 400 pts each, noted improvement in cognitive function, memory executive function and global outcome. The average BHB concentration in the Avgerinos study was 355 umol/l, representing a mild but significant increase compared to placebo/baseline.
Aside - A meta-analysis is a statistical analysis that combines the results of multiple scientific studies
In one of the largest randomized controlled trials to date, Henderson et al examined the impact of an MCT formulation (composed of glycerine and caprylic acid called AD-1202) on cognitive function in 152 patients with mild to moderate AD. The study ran for 90 days. Patients in the treatment arm had a significant improvement in cognitive test scores compared with placebo. BHB concentrations averaged 400 umol/l at day 90. The main benefit was noted in patients who were APO e4(-). The main side effect was GI intolerance.
The only evidence to date for the use of ketone ester formulations comes from a case report by Newport et al (2015) of a 63-year-old male with severe AD who was started on KME (28.7 g, 3 times per day for 20 weeks). The family noted significant improvement in cognitive function over the trial period. Interestingly, this was most notable at the peaks of BHB concentration after each dose which ranged from 3 – 7 mmol/l, one hour after receiving the KME. He was also APO e4(+) which contradicts the Henderson study. The KME was well tolerated.

Beta-Hydroxybutyrate (BHB) concentrations rose to 3 to 7 mM 1 hour after ingestion of ketone monoester (KME) in three different doses, 25, 35, and 50 g, taken on separate days. The peak levels measured are in the range of those obtained during adherence to the classical ketogenic diet and are about 10-fold the concentrations achievable by medium-chain triglyceride (MCTG) administration. The findings suggest that substantially elevated blood ketone concentrations can be maintained throughout the day if KME is taken every 3 to 4 hours.
(Newport, Mary T., Theodore B. VanItallie, Yoshihiro Kashiwaya, Michael Todd King, and Richard L. Veech. “A New Way to Produce Hyperketonemia: Use of Ketone Ester in a Case of Alzheimer’s Disease.” Alzheimer’s & Dementia 11, no. 1 (January 2015): 99–103. https://doi.org/10.1016/j.jalz.2014.01.006.)
Wrapping it all up
AD is a fairly common and always devastating neurological disease which appears at least partly, to result from a failure of cerebral energetics. As such, the improvements in cognitive function demonstrated by the various ketone studies appears to make physiological sense, as ketones provide an excellent energy substrate for the brain. The fact that glucose hypometabolism predates the onset of clinical AD, provides further impetus to supplementing cerebral energy substrates. Although we’ve seen a number of positive studies with exogenous ketone supplementation, a large, multicentre RCT is really required to once and for all, confirm (or refute) the benefit of ketone supplementation in AD. The best method of achieving ketosis in this setting remains to be determined.
References
Augustin, K., Khabbush, A., Williams, S., Eaton, S., Orford, M., Cross, J. H., Heales, S. J. R., Walker, M. C., & Williams, R. S. B. (2018). Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. The Lancet Neurology, 17(1), 84–93. https://doi.org/10.1016/S1474-4422(17)30408-8
Avgerinos, K. I., Egan, J. M., Mattson, M. P., & Kapogiannis, D. (2020). Medium Chain Triglycerides induce mild ketosis and may improve cognition in Alzheimer’s disease. A systematic review and meta-analysis of human studies. Ageing Research Reviews, 58, 1. https://doi.org/10.1016/j.arr.2019.101001
Broom, G. M., Shaw, I. C., & Rucklidge, J. J. (2019). The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition, 60, 118–121. https://doi.org/10.1016/j.nut.2018.10.003
Castellano, C.-A., Nugent, S., Paquet, N., Tremblay, S., Bocti, C., Lacombe, G., Imbeault, H., Turcotte, É., Fulop, T., & Cunnane, S. C. (2015). Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. Journal of Alzheimer’s Disease, 43(4), 1343–1353.
Cunnane, S. C., Courchesne-Loyer, A., St-Pierre, V., Vandenberghe, C., Pierotti, T., Fortier, M., Croteau, E., & Castellano, C.-A. (2016). Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer’s disease: Brain glucose and ketone uptake in Alzheimer’s disease. Annals of the New York Academy of Sciences, 1367(1), 12–20. https://doi.org/10.1111/nyas.12999
Fortier, M., Castellano, C.-A., Croteau, E., Langlois, F., Bocti, C., St-Pierre, V., Vandenberghe, C., Bernier, M., Roy, M., Descoteaux, M., Whittingstall, K., Lepage, M., Turcotte, É. E., Fulop, T., & Cunnane, S. C. (2019). A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimer’s & Dementia, 15(5), 625–634. https://doi.org/10.1016/j.jalz.2018.12.017
Gasior, M., Rogawski, M. A., & Hartman, A. L. (2006). Neuroprotective and disease-modifying effects of the ketogenic diet. Behavioural Pharmacology, 17(5–6), 431.
Grammatikopoulou, M. G., Goulis, D. G., Gkiouras, K., Theodoridis, X., Gkouskou, K. K., Evangeliou, A., Dardiotis, E., & Bogdanos, D. P. (2020). To Keto or Not to Keto? A Systematic Review of Randomized Controlled Trials Assessing the Effects of Ketogenic Therapy on Alzheimer Disease. Advances in Nutrition. https://doi.org/10.1093/advances/nmaa073
Guzmán, M., & Blázquez, C. (2004). Ketone body synthesis in the brain: Possible neuroprotective effects. Prostaglandins, Leukotrienes and Essential Fatty Acids, 70(3), 287–292. https://doi.org/10.1016/j.plefa.2003.05.001
Hashim, S. A., & VanItallie, T. B. (2014). Ketone body therapy: From the ketogenic diet to the oral administration of ketone ester. Journal of Lipid Research, 55(9), 1818–1826. https://doi.org/10.1194/jlr.R046599
Henderson, S. T. (2008). Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics, 5(3), 470–480. https://doi.org/10.1016/j.nurt.2008.05.004
Henderson, S. T., Vogel, J. L., Barr, L. J., Garvin, F., Jones, J. J., & Costantini, L. C. (2009). Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutrition & Metabolism, 6(1), 31. https://doi.org/10.1186/1743-7075-6-31
Hertz, L., Chen, Y., & Waagepetersen, H. S. (2015). Effects of ketone bodies in Alzheimer’s disease in relation to neural hypometabolism, β-amyloid toxicity, and astrocyte function. Journal of Neurochemistry, 134(1), 7–20. https://doi.org/10.1111/jnc.13107
Kashiwaya, Y., Takeshima, T., Mori, N., Nakashima, K., Clarke, K., & Veech, R. L. (2000). D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America, 97(10), 5440–5444.
Krikorian, R., Shidler, M. D., Dangelo, K., Couch, S. C., Benoit, S. C., & Clegg, D. J. (2012). Dietary ketosis enhances memory in mild cognitive impairment. Neurobiology of Aging, 33(2), 425.e19-425.e27. https://doi.org/10.1016/j.neurobiolaging.2010.10.006
L. Veech, Britton Chance, Yoshihiro, R. (2001). Ketone Bodies, Potential Therapeutic Uses. IUBMB Life (International Union of Biochemistry and Molecular Biology: Life), 51(4), 241–247. https://doi.org/10.1080/152165401753311780
Lilamand, M., Porte, B., Cognat, E., Hugon, J., Mouton-Liger, F., & Paquet, C. (2020). Are ketogenic diets promising for Alzheimer’s disease? A translational review. Alzheimer’s Research & Therapy, 12(1). https://doi.org/10.1186/s13195-020-00615-4
Maalouf, M., Rho, J. M., & Mattson, M. P. (2009). The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Research Reviews, 59(2), 293–315. https://doi.org/10.1016/j.brainresrev.2008.09.002
Mamelak, M. (2017). Energy and the Alzheimer brain. Neuroscience & Biobehavioral Reviews, 75, 297–313. https://doi.org/10.1016/j.neubiorev.2017.02.001
Morrill, S. J., & Gibas, K. J. (2019). Ketogenic diet rescues cognition in ApoE4+ patient with mild Alzheimer’s disease: A case study. Diabetes & Metabolic Syndrome: Clinical Research & Reviews, 13(2), 1187–1191. https://doi.org/10.1016/j.dsx.2019.01.035
Murray, A. J., Knight, N. S., Cole, M. A., Cochlin, L. E., Carter, E., Tchabanenko, K., Pichulik, T., Gulston, M. K., Atherton, H. J., Schroeder, M. A., Deacon, R. M. J., Kashiwaya, Y., King, M. T., Pawlosky, R., Rawlins, J. N. P., Tyler, D. J., Griffin, J. L., Robertson, J., Veech, R. L., & Clarke, K. (2016a). Novel ketone diet enhances physical and cognitive performance. The FASEB Journal, 30(12), 4021–4032. https://doi.org/10.1096/fj.201600773R
Murray, A. J., Knight, N. S., Cole, M. A., Cochlin, L. E., Carter, E., Tchabanenko, K., Pichulik, T., Gulston, M. K., Atherton, H. J., Schroeder, M. A., Deacon, R. M. J., Kashiwaya, Y., King, M. T., Pawlosky, R., Rawlins, J. N. P., Tyler, D. J., Griffin, J. L., Robertson, J., Veech, R. L., & Clarke, K. (2016b). Novel ketone diet enhances physical and cognitive performance. The FASEB Journal, 30(12), 4021–4032. https://doi.org/10.1096/fj.201600773R
Neth, B. J., Mintz, A., Whitlow, C., Jung, Y., Solingapuram Sai, K., Register, T. C., Kellar, D., Lockhart, S. N., Hoscheidt, S., Maldjian, J., Heslegrave, A. J., Blennow, K., Cunnane, S. C., Castellano, C.-A., Zetterberg, H., & Craft, S. (2020). Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: A pilot study. Neurobiology of Aging, 86, 54–63. https://doi.org/10.1016/j.neurobiolaging.2019.09.015
Newport, M. T., VanItallie, T. B., Kashiwaya, Y., King, M. T., & Veech, R. L. (2015). A new way to produce hyperketonemia: Use of ketone ester in a case of Alzheimer’s disease. Alzheimer’s & Dementia, 11(1), 99–103. https://doi.org/10.1016/j.jalz.2014.01.006
Norwitz, N. G., Jaramillo, J. G., Clarke, K., & Soto, A. (2020). Ketotherapeutics for neurodegenerative diseases. In International Review of Neurobiology (Vol. 155, pp. 141–168). Elsevier. https://doi.org/10.1016/bs.irn.2020.02.003
Ota, M., Matsuo, J., Ishida, I., Takano, H., Yokoi, Y., Hori, H., Yoshida, S., Ashida, K., Nakamura, K., Takahashi, T., & Kunugi, H. (2019). Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neuroscience Letters, 690, 232–236. https://doi.org/10.1016/j.neulet.2018.10.048
Paoli, A., Bianco, A., Damiani, E., & Bosco, G. (2014). Ketogenic Diet in Neuromuscular and Neurodegenerative Diseases. BioMed Research International, 2014, 1–10. https://doi.org/10.1155/2014/474296
Reger, M. A., Henderson, S. T., Hale, C., Cholerton, B., Baker, L. D., Watson, G. S., Hyde, K., Chapman, D., & Craft, S. (2004). Effects of β-hydroxybutyrate on cognition in memory-impaired adults. Neurobiology of Aging, 25(3), 311–314. https://doi.org/10.1016/S0197-4580(03)00087-3
Rho, J. M., & Stafstrom, C. E. (2012). The ketogenic diet as a treatment paradigm for diverse neurological disorders. Neuropharmacology, 3, 59. https://doi.org/10.3389/fphar.2012.00059
Stafstrom, C. E., & Rho, J. M. (2012). The ketogenic diet as a treatment paradigm for diverse neurological disorders. Frontiers in Pharmacology, 3, 59. https://doi.org/10.3389/fphar.2012.00059
Taylor, M. K., Swerdlow, R. H., Burns, J. M., & Sullivan, D. K. (2019). An Experimental Ketogenic Diet for Alzheimer Disease Was Nutritionally Dense and Rich in Vegetables and Avocado. Current Developments in Nutrition, 3(4). https://doi.org/10.1093/cdn/nzz003
Taylor, M. K., Swerdlow, R. H., & Sullivan, D. K. (2019). Dietary Neuroketotherapeutics for Alzheimer’s Disease: An Evidence Update and the Potential Role for Diet Quality. Nutrients, 11(8), 1910. https://doi.org/10.3390/nu11081910
Torosyan, N., Sethanandha, C., Grill, J. D., Dilley, M. L., Lee, J., Cummings, J. L., Ossinalde, C., & Silverman, D. H. (2018). Changes in regional cerebral blood flow associated with a 45 day course of the ketogenic agent, caprylidene, in patients with mild to moderate Alzheimer’s disease: Results of a randomized, double-blinded, pilot study. Experimental Gerontology, 111, 118–121. https://doi.org/10.1016/j.exger.2018.07.009
Ulamek-Koziol, M., & Pluta, R. (2020). To treat or not to treat Alzheimer’s disease by the ketogenic diet? That is the question. Neural Regeneration Research, 15(5), 857. https://doi.org/10.4103/1673-5374.268900
VanItallie, T. B. (2015). Biomarkers, ketone bodies, and the prevention of Alzheimer’s disease. Metabolism, 64(3), S51–S57. https://doi.org/10.1016/j.metabol.2014.10.033
VanItallie, T. B. (2017). Alzheimer’s disease: Innate immunity gone awry? Metabolism, 69, S41–S49. https://doi.org/10.1016/j.metabol.2017.01.014
Xie, G., Tian, W., Wei, T., & Liu, F. (2015). The neuroprotective effects of β-hydroxybutyrate on Aβ-injected rat hippocampus in vivo and in Aβ-treated PC-12 cells in vitro. Free Radical Research, 49(2), 139–150. https://doi.org/10.3109/10715762.2014.987274
Yin, J. X., Maalouf, M., Han, P., Zhao, M., Gao, M., Dharshaun, T., Ryan, C., Whitelegge, J., Wu, J., Eisenberg, D., Reiman, E. M., Schweizer, F. E., & Shi, J. (2016). Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiology of Aging, 39, 25–37. https://doi.org/10.1016/j.neurobiolaging.2015.11.018
Comments